SAEDNEWS: For the first time in history, doctors successfully used personalized CRISPR-based gene editing to treat a newborn with a rare and fatal genetic disorder (CPS1 deficiency). This groundbreaking intervention marks a major step toward individualized therapies for rare genetic diseases.
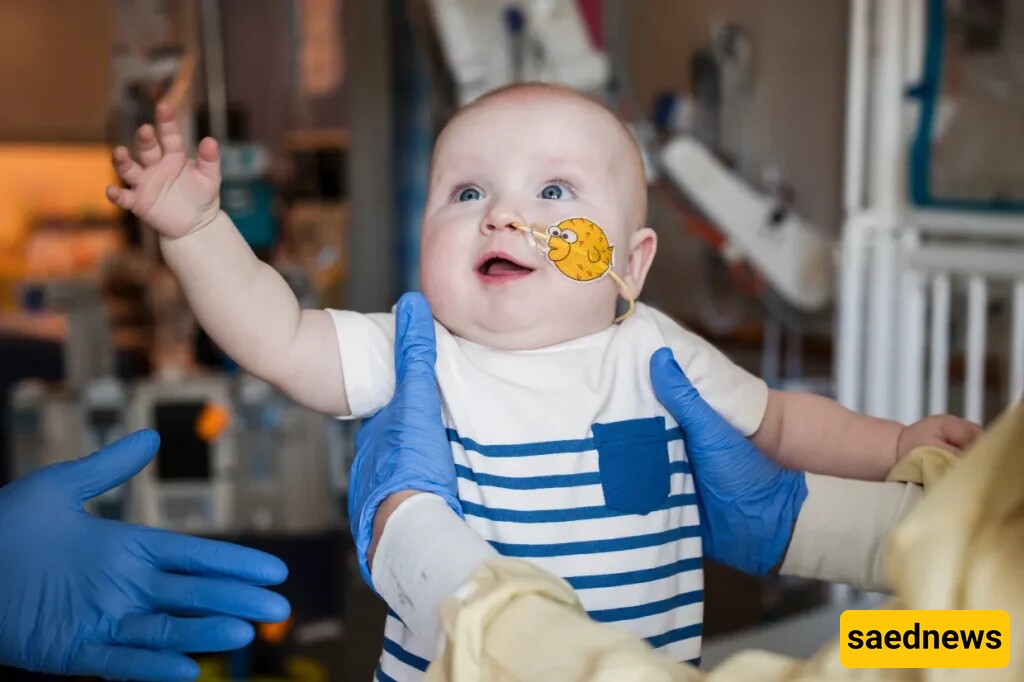
According to SAEDNEWS, In an unprecedented medical milestone, a team of physicians and scientists at the Children’s Hospital of Philadelphia (CHOP) has successfully rewritten the DNA of a baby suffering from a rare and fatal genetic condition — marking the first time in history that gene editing has been used to treat a newborn in this way.
The patient, 15-month-old KJ Muldoon, was diagnosed with CPS1 deficiency, a rare genetic disorder that disrupts the body’s ability to remove excess ammonia from the bloodstream. This condition leads to a rapid and dangerous buildup of ammonia that can cause severe brain damage or death if left untreated. The disease affects only 1 in 800,000 to 1.3 million births.
Shortly after KJ’s birth, his parents Nicole and Kyle noticed alarming symptoms, including lethargy and breathing difficulties. Medical tests revealed critically high levels of ammonia in his blood, confirming a diagnosis of Carbamoyl Phosphate Synthetase 1 (CPS1) deficiency, one of the most severe forms of urea cycle disorders.
Nicole described her reaction in a video released by CHOP:
“When you Google CPS1 deficiency, what you see is death statistics and liver transplants. We were completely devastated.”

Initially, doctors placed KJ on dialysis to reduce ammonia levels and stabilize his condition. However, traditional treatment methods, including liver transplant, were not suitable due to his frailty. That’s when the CHOP team proposed something radical: an experimental gene-editing therapy customized specifically for KJ.
Dr. Rebecca Ahrens-Nicklas, a pediatric geneticist leading the case, said:
“The biggest concern was not to give the family false hope. But we reached a point where we thought maybe there really was a team capable of designing a drug just for KJ.”
The drug, developed in partnership with scientists at the University of Pennsylvania, used the revolutionary CRISPR gene-editing technology. CRISPR works like a molecular scalpel: it identifies a specific segment of faulty DNA and replaces it with a correct version. In KJ’s case, the team created a fully personalized treatment targeting the exact mutation responsible for his disorder.
Remarkably, they designed and manufactured the drug in just six months — an extraordinary feat for a treatment never used before in humans.
In February 2025, KJ received the first dose of the gene-editing drug via intravenous injection. The treatment traveled through his bloodstream and reached the liver, where guide molecules directed the CRISPR system to the precise location of the genetic error. The system then cut out the mutated DNA and replaced it with a healthy sequence.
Nicole explained:
“I think of gene editing like writing on paper — when you make a mistake in a sentence, you go back and fix it. That’s exactly what they did to his DNA.”
To minimize risk, doctors began with the lowest possible dose, giving KJ's body time to adjust. Just days later, his condition began to improve. His ammonia levels stabilized, he could consume more protein without adverse effects, and his energy and physical development improved rapidly.
Over the next two months, he received two additional doses, each slightly stronger. He continues to be monitored at CHOP, and no serious side effects have been reported.
“We lived in fear for so long, but now we’re starting to see light at the end of the tunnel,” said Nicole. “From a frail little newborn to a strong, chubby, smiling baby — I’m glad we never gave up.”

While scientists are cautiously optimistic, they emphasize that it’s still too early to declare the treatment a definitive cure. KJ’s condition will continue to be monitored over time to assess the long-term safety and effectiveness of the gene-editing approach.
Nevertheless, the implications are enormous. If successful, this case could pave the way for hundreds of similar treatments for rare genetic and metabolic liver diseases. Dr. Ahrens-Nicklas hopes that children like KJ can one day live normal lives with minimal or even no medication.
“Our dream is to reach a point where, given a patient’s genetic mutation, we can rapidly design and deliver a treatment tailored just for them.”
Professor Gemma Marfany, a geneticist at the University of Barcelona who was not involved in the study, praised the innovation:
“This is the first case of a completely personalized gene-editing therapy for a newborn with an ultra-rare and severe disorder. It’s a remarkable proof of concept — science at its finest.”
This landmark achievement not only brings hope to one family, but also signals a future in which precision medicine becomes the norm, not the exception. The success of KJ’s treatment is more than a medical first — it may very well be the dawn of a new era in curing genetic diseases.







![Mysterious Ghost Captured by James Webb Telescope in Space [Photo]](https://en.saednews.com/storage/files/post/b9941a9d-7162-4b40-8090-ecfcc8b7ef59-GSffBk6i9vZEGrJX/1x1-image.jpg)